Artikelen
Artikelen - Gelokaliseerde sclerodermie bij kinderen (2019-03)
M.M.B. Seyger
Gelokaliseerde sclerodermie behelst een spectrum van sclerotische ziektebeelden waarbij primair de huid is aangedaan. Afhankelijk van het subtype zijn ook het onderhuidse vet, de fascie, spieren en zelfs het bot bij het proces betrokken. In tegenstelling tot systemische sclerodermie is er echter geen betrokkenheid van de interne organen. [1]
Er is enige onduidelijkheid over de naamgeving van het ziektespectrum. Hoewel in de Verenigde Staten de term morfea gehanteerd wordt opdat geen verwarring kan ontstaan met het ziektebeeld systemische sclerose, gebruiken veel andere (Europese) landen gelokaliseerde sclerodermie als overkoepelende term, omdat morfea gezien wordt als een subtype in het ziektespectrum. [2] Bovendien gebruiken kinderdermatologen en kinderreumatologen (ook de Amerikaanse) liever de term gelokaliseerde sclerodermie omdat het subtype lineaire sclerodermie het meest bij kinderen voorkomt (figuur 1). [3] In dit artikel zal de term juveniele gelokaliseerde sclerodermie (JLS) gebruikt worden. De incidentie van JLS is 0,3-3 per 100.000 kinderen en de gemiddelde leeftijd van ontstaan is tussen 7 en 9 jaar. Het komt veel vaker voor bij meisjes dan bij jongens (ratio 2,4:1). [3] Het type lineaire sclerodermie wordt veruit het meest gezien bij kinderen (65%), gevolgd door het plaque type of een combinatie van lineaire sclerodermie en plaque type morfea (mixed subtype). [4]
Classificatie
Met betrekking tot de classificatie van gelokaliseerde sclerodermie is eveneens geen complete consensus in de literatuur. De in 2017 verschenen European Dermatology Forum S1 guideline gebruikt de door Kreuter voorgestelde classificatie, waarin ook types als gutatta morphea, atrofoderma van Pierini en Pasini en eosinofiele fasciitis (schulmansyndroom) zijn opgenomen. [1] De classificatie van Laxer en Zulian wordt eveneens vaak gehanteerd [2] en wordt bovendien het meest toegepast in de kinderliteratuur. [4] In deze classificatie zijn vijf types opgenomen: circumscribed morfea, lineaire sclerodermie, gegeneraliseerde morfea, pansclerotische morfea en het mixed subtype (tabel 1).
De betrokkenheid van de huid in het type circumscribed superficiële morfea, ook wel plaque type morfea genoemd, beperkt zich tot de dermis. De ovale tot ronde laesies zijn vooral aanwezig op de romp. Deze vorm komt het meest voor bij volwassenen. Bij circumscribed diepe morfea (morphea profunda) reikt de sclerose tot de subcutis of zelfs tot de fascie en spieren. Dit type komt zowel bij kinderen als volwassenen weinig voor. [2,4]
Lineaire sclerodermie wordt het meest gezien bij kinderen en adolescenten. Alle lagen van de extremiteiten (tot aan het bot) kunnen betrokken zijn; meestal is de ziekte unilateraal aanwezig. Als de lineaire sclerodermie zich op het hoofd bevindt, wordt deze sclerodermie en coup de sabre (slag van het zwaard) genoemd. Meestal bevinden de laesies zich paramediaan op het voorhoofd, met veelal uitbreiding naar de scalp, waardoor alopecie op kan treden. Een speciale subvorm van lineaire sclerodermie van het hoofd is de progressieve hemifaciale atrofie (Parry Romberg). Opvallend bij deze vorm is het feit dat de huid van het gelaat niet of nauwelijks is aangedaan, maar er sprake is van unilateraal verlies van subcutis, spier en zelfs bot. Omdat in de meerderheid van de gevallen een sclerodermie en coupe de sabre en een Parry Romberg samen voorkomen, gaat men ervan uit dat beide tot hetzelfde ziektespectrum behoren. Gegeneraliseerde morfea en pansclerotische morfea komen beide zeer weinig voor bij kinderen. [2,4]
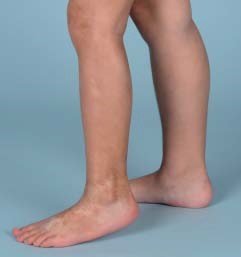
Figuur 1. Lineaire sclerodermie.
Ziekteactiviteit
Het monitoren van ziekteactiviteit en ziekteschade is een uitdaging bij de behandeling van JLS. Op dit moment wordt de Localized Scleroderma Cutaneous Assessment Tool (LoSCAT) het meest gebruikt als uitkomstmaat bij volwassenen, en in 2018 is deze ook gevalideerd voor het gebruik bij kinderen. [5] De LoSCAT verdeelt het lichaam in achttien regio’s en bestaat uit twee onderdelen: de modified Localized Scleroderma Skin Severity Index (mLoSSI) en de localized Scleroderma Skin Damage Index (LoSDI). De mLoSSI beoordeelt tekenen van ziekteactiviteit (uitbreiding van ziekte, aanwezigheid van erytheem en induratie). De LosDI schat de mate van ziekteschade in (cutane en subcutane atrofie, en pigmentveranderingen). [2] Hoewel de LoSCAT het behandeleffect goed kan meten, wordt er niet naar de kenmerken van een solitaire laesie gekeken als maat voor activiteit. Eind 2018 publiceerden Li et al. nieuwe kenmerken (van een solitaire laesie) die sterk geassocieerd zijn met ziekteactiviteit in JLS: erytheem, livide rand, warm aanvoelende laesie, abnormale huidtextuur (abnormaal glad, glanzend of wasachtig) en uitbreiding van de laesie. [6] Deze kenmerken kunnen een praktisch handvat bieden om de ziekteactiviteit van JLS te boordelen in de dagelijkse klinische praktijk.
Verschillen kinderen en volwassenen
Een groot verschil tussen sclerodermie bij kinderen en volwassenen is de uiteindelijke restschade, die bij kinderen meestal veel groter is. Omdat lineaire sclerodermie (zowel aan de extremiteiten als in het gelaat) bij kinderen veel vaker voorkomt, en het beloop hiervan vaak ernstiger is, worden contracturen, spieratrofie, verschil in beenlengte- en omvang, faciale hemiatrofie en ernstige misvorming veel vaker bij kinderen gezien.
[6] Bovendien staat de lineaire vorm van sclerodermie bekend als een chronische ziekte die vaak recidiveert. In een recente retrospectieve analyse beschrijven Mertens et al. dat van de patiënten met een gelokaliseerde sclerodermie die is ontstaan op kinderleeftijd, 27% een recidief krijgt. Het lineaire type is een extra risicofactor voor het krijgen van een recidief. Recidieven treden gemiddeld op na twee jaar, maar kunnen zelfs na twintig jaar nog voorkomen. [7]
Bij kinderen worden vaker extracutane verschijnselen gerapporteerd, die eveneens het meest voorkomen bij het lineaire subtype. Terwijl artritis en artralgieën genoemd worden bij lineaire sclerodermie van de extremiteiten, worden neurologische problemen (hoofdpijn, epilepsie en gedragsveranderingen), oogproblemen (met name uveitis) en odontostomatologische complicaties (abnormale dentitie, hypoplasie van mandibula en maxilla) beschreven bij lineaire sclerodermie en coup de sabre en bij progressieve faciale hemiatrofie van Parry Romberg. [4,8]
Behandeling
De behandeling van JLS zou met name gebaseerd moeten zijn op het type sclerodermie en de mate van activiteit. De oppervlakkige, niet-progressieve vorm van het plaque type morfea kan behandeld worden met topische middelen zoals corticosteroïden, calcineurineremmers, calcipotriol of een combinatie. Fototherapie met UVA-1 zou een alternatief kunnen zijn voor kinderen ouder dan 12 jaar met kleine superficiële, niet-pro-gressieve laesies die niet boven gewrichten zitten (superficiële plaque type). Alle andere types van JLS (lineaire sclerodermie, gegeneraliseerde of diepe morfea) dienen behandeld te worden met systemische immunosuppressie. [3,4] Eerste keus behandeling is methotrexaat in een dosering van 15 mg/m2/week (oraal of subcutaan) met een maximumdosering van 25 mg/ week. De eerste drie maanden is het aan te raden om ter overbrugging eveneens te behandelen met een afbouwschema prednison. Gezien het grote aantal recidieven wordt geadviseerd om patiënten indien mogelijk gedurende minstens twee jaar met methotrexaat te behandelen. [4] Indien methotrexaat ineffectief is, of er te veel bijwerkingen optreden, is mycofenolaat mofetil in een dosering van 750-1200 mg/m2/dag met een maximum van 2000-2500 mg/dag, een goed alternatief. [4,9]. Bij zeer refractaire vormen van JLS lijkt behandeling met tocilizumab hoopvol. [10]
Tabel 1. Classificatie van gelokaliseerde sclerodermie volgens Laxer en Zulian. Gemodificeerd uit Mertens et al. [2] 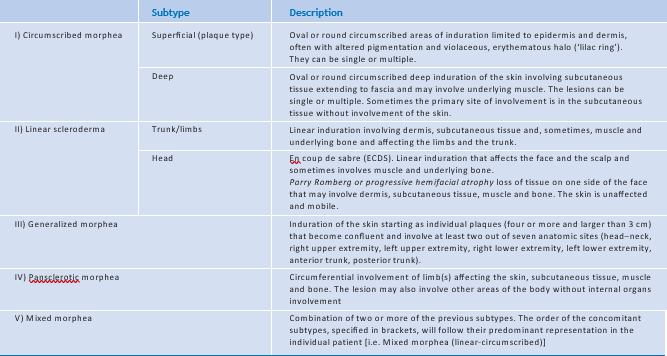
Extracutane manifestaties
De behandeling van JLS (zeker de lineaire en diepe vormen) gebeurt bij voorkeur op een gecombineerd spreekuur van een (kinder)reumatoloog en (kinder)dermatoloog. [1,3] Vanwege de eerder genoemde functionele en cosmetische deformiteiten en extracutane manifestaties, is het bij kinderen van groot belang om de diagnose snel te stellen en in de actieve fase van de ziekte adequaat te behandelen, om de kans op restverschijnselen te verminderen. [3,4] Naast de eerder genoemde medicamenteuze behandeling van JLS is het belangrijk aandacht te hebben voor de extracutane manifestaties van de ziekte. Bij kinderen met musculoskeletale betrokkenheid dient op artritis gescreend te worden en is verwijzing naar een fysiotherapeut belangrijk. Bij een dreigend beenlengteverschil is het noodzaak op tijd te verwijzen naar een (kinder)orthopeed, zodat de groei van het gezonde been indien nodig geremd kan worden. Kinderen met een lineaire sclerodermie en coup de sabre en/of progressieve faciale hemiatrofie wordt een MRI cerebrum geadviseerd. [4] Bij afwijkingen op de MRI en/of neurologische klachten is verwijzing naar de neuroloog geïndiceerd. Verder wordt, vooral bij dit type, screening op uveitis/oogheelkundige betrokkenheid geadviseerd. [3]
Bij odontostomatologische complicaties is verwijzing naar de mond-kaak-aangezichtschirurg aan te bevelen. [1,3] Tot slot dient de impact van JLS op de kwaliteit van leven niet onderschat te worden. [3] Het aanbieden van psychologische hulp en/of verwijzing naar de patiëntenvereniging KAISZ (Vereniging voor Kinderen met een Auto-Immuun of Auto-Inflammatoire Systeemziekte) kan hierbij helpen. Gezien het grote risico op recidieven is langdurige follow-up na succesvolle behandeling sterk aan te raden. [1,4]
Sinds 2018 is het Radboudumc een NFU erkend expertisecentrum voor zeldzame aandoeningen voor kinderen en volwassenen met gelokaliseerde sclerodermie, eosinofiele fasciitis en systemische sclerose. Binnen dit centrum worden kinderen met gelokaliseerde sclerodermie gezien op een gecombineerd spreekuur van de kinderreumatoloog en (kinder)dermatoloog. Middels duidelijk gedefinieerde zorgpaden worden patiënten indien nodig verwezen naar de kinderorthopeed, de mond- kaak-aangezichtschirurg, oogarts of de kinderneuroloog.
Literatuur
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European dermatology forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, Part 1: localized scleroderma, systemic sclerosis and overlap syndromes. J Eur Acad Dermatol Venereol 2017;31(9):1401-24.
- Mertens JS, Seyger MMB, Thurlings RM, Radstake TRDJ, de Jong EMGJ. Morphea and eosinophilic fasciitis: An update. Am J Clin Dermatol 2017;18(4):491-512.
- Constantin T, Foeldvari I, Pain CE, et al. Development of minimum standards of care for juvenile localized scleroderma. Eur J Pediatr 2018;177(7):961-77.
Correspondentieadres
Marieke Seyger
E-mail: marieke.seyger@radboudumc.nl