Artikelen
Artikelen - Rare knakkers: zeldzame huidtumoren (2019-03)
M.W. Bekkenk, Y.S. Elshot, S.E. Uitentuis, B. Zupan-Kajcovski, D. Tio
Naast de zeer frequent voorkomende basaalcelcarcinomen en plaveiselcelcarcinomen worden incidenteel ook andere typen niet-melanoom huidkanker geconstateerd. Denk hierbij aan het merkelcelcarcinoom (MCC), adnexcarcinomen, bepaalde types cutane lymfomen en allerlei soorten sarcomen van de huid. Bijna bij al deze tumoren wordt er initieel niet tot nauwelijks gedacht aan deze aandoeningen. [1,2] Bovendien komen veel van de zeldzame vormen van huidkanker vooral voor bij mensen die al bekend zijn met ‘gewone’ vormen van huidkanker, zoals bij solide orgaantransplantatiepatiënten en mensen met ernstig actinisch beschadigde huid. Toch is het belangrijk aan deze tumoren te denken, omdat het gedrag van veel van deze vormen van huidkanker anders is en veelal agressiever. Zodoende is verder onderzoek en aanvullende therapie regelmatig geïndiceerd.
Merkelcelcarcinoom
Het MCC is een vrij zeldzame, maar vaak agressieve tumor. Het presenteert zich meestal als een aspecifieke roodpaarse, snel groeiende, maar zelden ulcererende, laesie in het hoofd- halsgebied bij ouderen met een licht huidtype. Meer atypische presentaties, zoals cysteuze of subcutane laesies zijn beschreven. In Nederland worden ongeveer 170 nieuwe MCC’s geconstateerd per jaar, maar de incidentie stijgt snel. [3] In vergelijking: dat is ongeveer 35 keer minder vaak dan het melanoom. Na een biopt is de diagnose tegenwoordig relatief eenvoudig te stellen. Vooral de CK20, die eenduidig positief is, zorgt voor een meer eenduidige diagnose. [4] Overigens is de voorloper- cel van het MCC wel een groot discussiepunt. Aanvankelijk werd aan een neuro-endocriene oorsprong (de merkelcel) gedacht, dat zorgde voor de naam, maar de laatste tijd wordt dit steeds onwaarschijnlijker geacht. [5] Eén van de andere kandidaten is een stamcel van de dermale huid afkomstig uit de neurale lijst. Daarnaast is vanwege de positiviteit van de cellen voor paired box 5, ook een vroege B-cel genoemd als cel van oorsprong. [6] De rol van het merkelcelpolyoomvirus dat in ongeveer de helft van de MCC’s wordt gevonden is nog niet volledig opgehelderd, maar virusnegatieve gevallen blijken een slechtere prognose te hebben. Na de pathologische bevestiging van een MCC zal er extra aandacht moeten zijn voor de drainerende lymfklieren en bij verdenking wordt een echo met cytologische punctie geadviseerd. Er zal een schildwacht- klierprocedure (SWKP) moeten worden aangeboden indien er geen palpabele klieren aanwezig zijn. Het percentage dat na een SWKP positieve klieren blijkt te hebben is aanzienlijk: 25-30% van de patiënten met klinisch negatieve klieren blijkt (micro)metastasen in de SWK te hebben. [7] Aangezien het bijna altijd oudere patiënten betreft met laesies in het hoofd- halsgebied (waar bekend is dat de SWKP minder sensitief is en meer kans geeft op morbiditeit) en de impact op de prognose van het verwijderen van deze klieren niet geheel duidelijk is, moet deze ingreep goed besproken worden met de patiënt. Bij twijfel over metastasen op afstand kan een (pet-)CT-scan worden overwogen. De prognose van het MCC is matig, zelfs bij presentatie met alleen lokale ziekte is er een vijfjaarsoverleving van rond de 50%, maar dat zakt naar 15% bij al aanwezige metastasen bij diagnose. [8] De belangrijkste therapie is chirurgie, waarbij er internationaal discussie is over de vrije marge, waarbij tot 2 cm marge wordt geadviseerd. Bij een recente systematische review van de literatuur lijkt er geen verschil in de prognose te ontstaan na excisie door Mohs, met 1 cm of met 2 cm marge. [9] Radiotherapie lijkt een goede (additionele) therapie, ook de drainerende lymfklieren zouden bij een positieve SWKP mee bestraald kunnen worden. Primaire radiotherapie kan overwogen worden bij patiënten die geen kandidaten zijn voor chirurgie. [10] Voor het gemetastaseerde MCC is chemotherapie weinig effectief, immunotherapie (door middel van anti-PD1-therapie) is een interessante nieuwe optie, maar is nog maar bij een zeer beperkt aantal patiënten ingezet. [11]
Adnexcarcinomen
Deze heterogene groep tumoren, die afkomstig is van de hui- dadnexen, presenteert zich vaak in het hoofd-halsgebied. Het is een zeldzame groep, met een geschat aantal nieuwe gevallen per jaar in Nederland rond de tachtig en vooral bij mensen op hoge leeftijd. Ook voor deze groep geldt dat er zelden initieel gedacht wordt aan een maligne adnextumor. [1] Deze tumoren hebben geen specifieke klinische kenmerken, kunnen op andere soorten niet-melanoom kanker lijken en omdat ze zeldzaam voorkomen kunnen zowel de clinicus als de patholoog hiermee moeilijk ervaring opdoen. Daarom is ook de histologische diagnose soms (zeer) lastig. Toch is het belang van duidelijke onderlinge differentiatie en juiste diagnose groot omdat de kans op (lokale) recidieven en metastasering ver- schilt. Daarnaast moet er ook gedacht worden dat sommige van deze adnexcarcinomen in het kader van bepaalde erfelijke aandoeningen kunnen voorkomen, zoals talgkliercarcinomen in Llynch/Muir-Torre en spiradenocarcinomen in brooke- spieglersyndroom.
Een van de meest voorkomende uit de groep apocriene- eccriene carcinomen (figuur 1) is het microcysteus adnexcarcinoom dat notoir lastig kan zijn om lokaal volledig te verwijderen. Vanwege hoge recidiefkans (> 50%!) wordt mohschirurgie dan ook regelmatig geadviseerd. Hierbij is een belangrijke valkuil de verwarring met het benigne plaquetype syringoom dat kan leiden tot overbehandeling. [12]
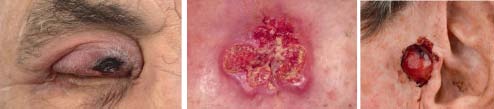
Figuur 1. Lokalisatie cutaan T-cellymfoom, eccrien carcinoom en een pleiomorf sarcoom.
Sarcomen
Deze heterogene groep van tumoren bevat een aantal entiteiten die niet alleen een verschillende presentatie hebben, maar ook een zeer verschillende prognose hebben en behandeling behoeven. De meest bekende en relatief frequent voorkomende vorm van het sarcoom: het dermatofibrosarcoma protuberans (DFSP) wordt vaak initieel gemist. Deze aandoening oogt vaak initieel als een dermatofibroom of verlittekening en treft ook jongere personen en zelfs kinderen, zodat er vaak niet direct aan maligniteit wordt gedacht. Deze tumor groeit langzaam en heeft een laag metastaserend potentieel. De behandeling is dan ook vooral chirurgisch, waarbij mohschirurgie mogelijk minder kans geeft op een lokaal recidief. [13]
Er is discussie of het atypisch fibroxanthoom (AFX) ook beschouwd moet worden als een type sarcoom. De afgrenzing tussen het AFX en het pleiomorf dermaal sarcoom (ook bekend als ongedifferentieerd pleiomorf sarcoom) (figuur 1) is niet eenvoudig en mogelijk betreft het een spectrum waarbij bij het AFX de maligne cellen meer oppervlakkig en bij het onge- differentieerde pleiomorf sarcoom dieper gelegen zijn. Het betreft snel groeiende, rode (soms ulcererende) plaques of nodi die zich presenteren op (sterk) zonbeschadigde huid van vooral oudere personen, die vaak al bekend zijn met andere vormen van huidkanker. Chirurgie is de eerstekeustherapie, metastasering gebeurt relatief zeldzaam en is overwegend lymfogeen. Bij het pleiomorf dermaal sarcoom is er sprake van een hogere kans op metastasering, waarbij adjuvante radiotherapie, naast chirurgie, mogelijk een rol heeft. [14]
Angiosarcomen hebben een vrij specifieke presentatie, maar zowel klinisch als histologisch kunnen bij vroege tumoren de maligne kenmerken niet evident zijn en worden ze nogal eens beoordeeld als een benigne vasculaire aandoening. [15] De prognose van de angiosarcomen is slecht, waarschijnlijk mede door de vaak late diagnose. De metastasering verloopt over het algemeen hematogeen. Een beruchte presentatie is een angiosarcoom in een eerder bestraald gebied (vooral status na mammacarcinoom) en wordt dan ook het stewarttrevessyndroom genoemd. [16]
Andere sarcomen van de huid zijn zeer zeldzaam, het leiomyosarcoom, liposarcoom en andere vormen van het sarcoom van de huid zijn zo zeldzaam dat ze geen standaard beleid hebben en dat bespreking in een (sarcoom) multidisciplinair overleg wordt geadviseerd. Er zal bijna altijd een combinatie van chirurgie en radiotherapie worden geadviseerd, respons op chemotherapeutische behandeling is over het algemeen laag.
Cutane lymfomen (figuur 1)
Een aantal primaire cutane lymfomen presenteren zich met een solitaire laesie en kunnen klinisch makkelijk verward worden met bijvoorbeeld een sarcoom van de huid. Primair cutane lymfomen die zich kunnen presenteren met een solitaire laesie kunnen zowel van (NK-)T-celorigine als B-celo- rigine zijn. Voor de T-cellymfomen zijn het vooral het primair cutaan CD30+ grootcellig anaplastisch lymfoom, primair cutaan CD4+ klein/medium T-cellymfoproliferatie en het perifeer T-cellymfoom en voor de B-cellymfomen betreft het primair cutaan marginalezonelymfoom, het primair cutaan follikelcentrumlymfoom en het primair cutaan grootcellig
B-cellymfoom, leg-type. [17] Natuurlijk kan ook een nodaal lymfoom zich presenteren in de huid en zal er adequate stage- ring moeten plaatsvinden om te kunnen vaststellen dat het een primair cutaan lymfoom betreft. De prognose en therapie wisselt sterk tussen de verschillende subtypes primair cutane lymfomen, waarbij er soms een zeer goede prognose (CD4+ klein/medium T-cellymfoproliferatie, CD30+ grootcellig anaplastisch T-cellymfoom, marginale zone B-cellymfoom en follikelcentrumlymfoom), maar soms ook erg slecht (perifeer T-cellymfoom NOS).
Literatuur
- Stam H, van de Wiel BA, Klop WMC, et al. Skin adnexal carcinoma of the head and neck: a retrospective study in a tertiary referral center. Eur Arch Otorhinolaryngol 2015;272(4):1001-10.
- Pasta V, Vergine M, D’Orazi V, Scipioni P, Monti M, Redler A. Merkel-cell carcinoma: preoperative clinical diagnosis and therapeutic implicati- ons. Ann Ital Chir 2014;85(4):352-7.
- Uitentuis SE, et al. Treatment and survival of Merkel cell carcinoma since 1993: a population-based cohort study in the Netherlands. Submitted.
- Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carci- nomas. A new histodiagnostic marker detected by monoclonal anti bodies. Am J Pathol 1992;140(2):427-47.
- Tilling T, Moll I. Which are the cells of origin in merkel cell carcinoma? J Skin Cancer 2012;2012:680410.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res 2013;73(16):4982-7.
- Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 2005;23(10):2300-9.
- Harms KL, Healy MA, Nghiem P, et al. Analysis of prognostic factors from 9387 merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol 2016;23(11):3564-71.
- Uitentuis SE, et al. Merkel cell carcinoma, the impact of clinical exci- sion margins and Mohs micrographic surgery on recurrence and sur vival: a systematic review, submitted.
- Green MD, Hayman JA. Radiotherapy in the multidisciplinary management of merkel cell carcinoma. J Natl Compr Canc Netw. 2018;16(6):776-81.
- Sellah D, Saint-Jean M, Peuvrel L, Khammari A, Quereux G, Dreno B. Anti-PD1 in Merkel cell carcinoma and cutaneous squamous cell carcinoma, description of five cases and recent data from the litera- ture. J Eur Acad Dermatol Venereol 2018.
- Wallace JS, Bond JS, Seidel GD, Samie FH. An important mimicker: plaque-type syringoma mistakenly diagnosed as microcystic adnexal carcinoma. Dermatol Surg 2014;40(7):810-2.
- Veronese F, Boggio P, Tiberio R, et al. Wide local excision vs. Mohs Tubingen technique in the treatment of dermatofibrosarcoma protu- berans: a two-centre retrospective study and literature review. J Eur Acad Dermatol Venereol 2017;31(12):2069-76.
- Soleymani T, Tyler Hollmig S. Conception and management of a poorly understood spectrum of dermatologic neoplasms: Atypical fibroxanthoma, pleomorphic dermal sarcoma, and undifferentiated pleomorphic sarcoma. Curr Treat Options Oncol 2017;18(8):50.
- Shustef E, Kazlouskaya V, Prieto VG, Ivan D, Aung PP. Cutaneous angi- osarcoma: a current update. J Clin Pathol 2017;70(11):917-25.
- Sharma A, Schwartz RA. Stewart-Treves syndrome: pathogenesis and management. J Am Acad Dermatol 2012;67(6):1342-8.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cuta- neous lymphomas. Blood 2005;105(10):3768-85.

Correspondentieadres
Marcel Bekkenk
E-mail: m.w.bekkenk@amc.uva.nl